GeneValidator
GeneValidator
GeneValidator is a tool to identify problematic gene predictions based on comparisons between gene predictions and similar sequences in public databases (e.g., SwissProt). Funded by NESCent Google Summer of Code 2013 and BBSRC TRDF. It can be used from the command-line (for HTML or parseable .txt outputs) or using a web interface as described below.
Check the main Genevalidator site for more details
Mini-tutorial for interactive use on one or few gene predictions
Despite recent improvements in genome sequencing and gene prediction technologies, many gene predictions remain problematic. GeneValidator can be used to help assess the quality of a large set of gene predictions, but also for individual sequences. Here we focus on the latter.
- Take one or several gene predictions in FASTA format - protein (e.g., A., B., C.) or nucleotide sequence (e.g., D.).
- Go to the GeneValidator web app.
- Paste your gene prediction into the text field and click the Analyse Sequences button.
GeneValidator will BLAST your gene prediction against a database (default: SwissProt), and perform multiple comparisons between your gene prediction and the sequences in the database. This should take less than 2 minutes. - Examine the output report:
- GeneValidator will only report results if it identified sufficient similar sequences in the database.
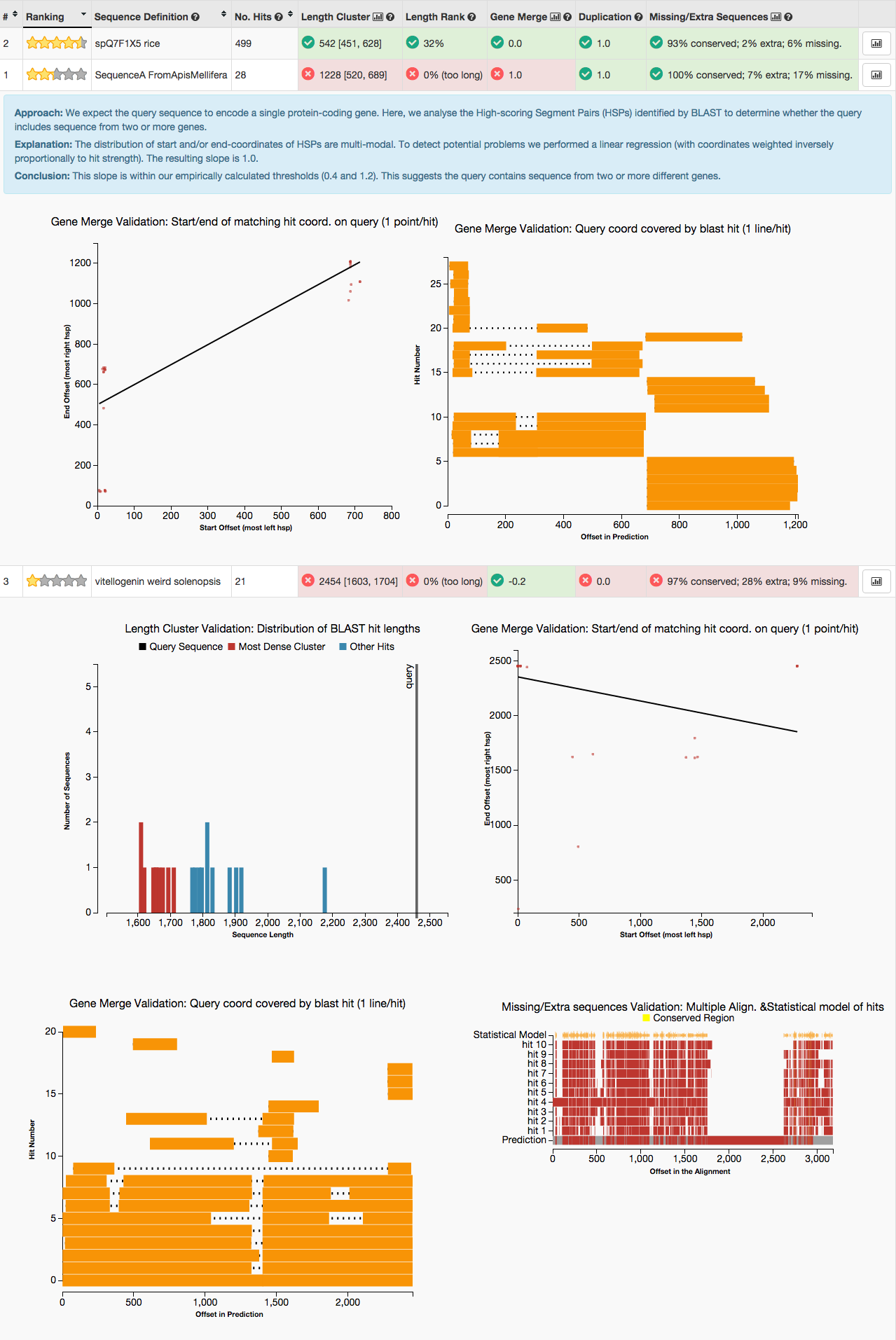
- Each test result is shown in a different column. The question mark buttons provide details about the test.
- Each test result is accompanied by an indication of consistency between the gene prediction and the BLAST hits.
- GeneValidator produces visual graphs to help understand the characteristics of the gene prediction data.
What can you conclude regarding your query gene prediction sequences? Regarding the example sequences given above, the following can help you understand GeneValidator's output:
- There appear to be several problems with this gene:
- It is longer than most BLAST hits (see Length Cluster graph)
- Each BLAST hit aligns either to the first part or to the second part of the query sequence (see second Gene Merge graph). This (along with the first Gene Merge graph) suggests the query may be a fusion of two genes (this happens occasionally for tandem genes).
- There is no evidence of any problems with this gene.
- A region of the gene aligns multiple times to a single BLAST hit as indicated by the duplication result. This suggests that our query gene prediction may include a single exon twice (e.g., as a result of prediction software incorrectly merging tandem (adjacent) duplicated gene copies into a single prediction.
- This sequence likely contains a frameshift. This is indicated by BLAST hits not all aligning in a single reading frame and by the presence of two main open reading frames.